| 《医疗器械定期风险评价报告》撰写要点 |
| 发布日期:2021/7/2 点击数:1185 次 新闻来源:CMD |
2018年8月31日,国家药品监督管理局发布《医疗器械不良事件监测和再评价管理办法》(国家市场监督管理总局令第1号)(下文简称1号令),它给医疗器械注册人提出了一系列新的工作要求,其中就包括对上市后的医疗器械撰写定期风险评价报告。本文就对在撰写该报告过程中可能遇到的常见问题做科普与解答。 要点整理  01 实施背景 根据1号令第二章第十四条"持有人的主要义务"的内容,持有人应做到建立体系、配备人员和机构、报告不良事件、控制风险、发布信息、撰写定期风险评价报告、再评价、配合调查。撰写定期风险评价报告时的基本注意事项和要求可以参照1号令第三十八 - 第四十一条的内容。 该报告与《医疗器械不良事件年度汇总报告表》相比,收集方法更丰富,收集范围更广泛,汇总方式更精确,分析内容更全面。 02 重要概念
03 撰写要点 - 总览
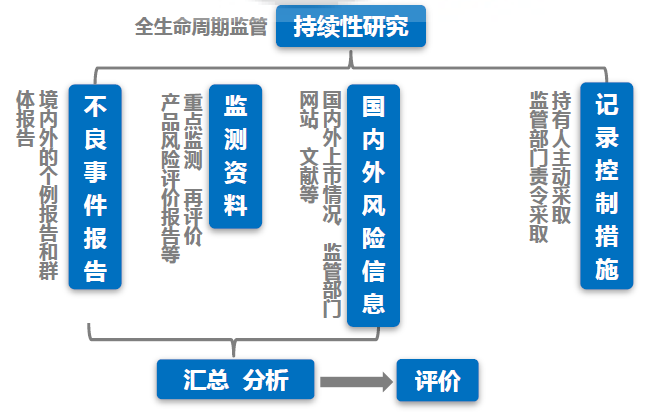
注意:即便没有发生不良事件,还是需要定期按时提交定期风险评价报告。不良事件报告是定期风险评价报告中的一部分,但不是全部内容。还有监测资料,国内外风险信息需要汇总和分析。
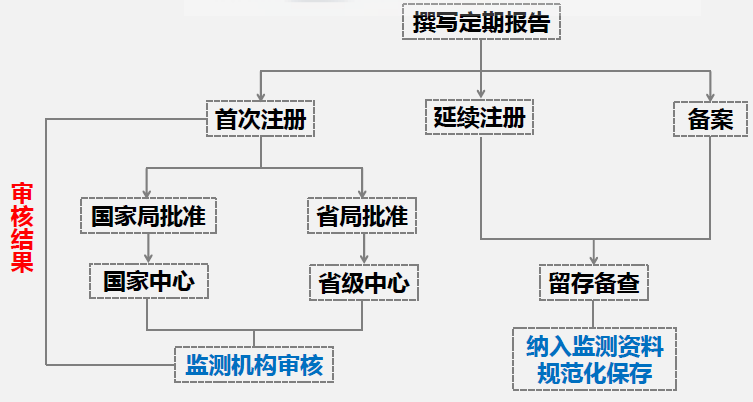
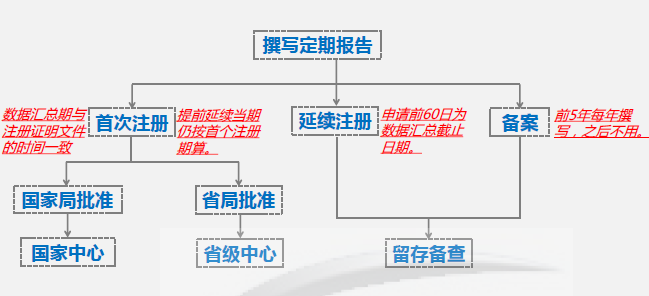
注意:对于首次注册的II, III类产品,网上提交资料后,依据审批机构的不同,会由国家局或省局进行审核,审核结果可在网上查询。对于延续注册和备案产品来说,不要求在线提交定期风险评价报告,撰写完成后留存备查即可,同时把定期风险评价报告纳入监测资料规范化进行保存。 04 撰写要点 - 正文内容
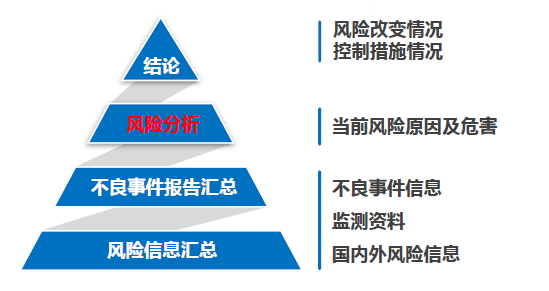
对于医疗器械产品,安全性有保证是重中之重。通过对产品的不良事件进行监测,定期分析产品的风险是保障产品安全的重要途径之一。因此,在撰写定期风险评价报告时,注册人应从识别风险和原因分析两方面撰写风险分析,评估其对安全性的影响。识别风险包括:对不良事件的特点、频率、伤害严重程度等进行分析;原因分析应从:设计、生产管理、流通与储存、使用因素等方面考虑。
在正文的结论部分,注册人应指出: 1. 指出本期报告与既往报告的风险分析结果差异 2. 指出以上风险差异的可接受程度 3. 总结采取的风险控制措施 05 撰写要点 - 时间节点
重点解读: 1. “数据汇总期与注册证明文件的时间一致” - 比如某个产品的获证日期是2018年8月10日,那么在2019年度需要提交的定期风险评价报告的数据汇总期就是2018年8月10日至2019年8月9日。 2. “提前延续当期扔按首个注册期算” - 比如某个产品2020年8月30日到期,但它在2020年5月就获得了延续注册证。对于该产品,本期报告仍以2020年8月30日作为汇总期的结束日期。
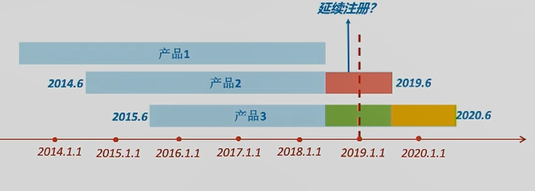
案例说明:
问:上图中3个II类产品提交定期风险评价报告时,数据汇总期应该分别是哪一段时间呢? 答:某II类产品1,其首个注册期是2013年某月某日到2018年某月某日。对这个产品来说,2019年1月1日1号令生效时,其处于延续注册状态,所以只需准备下一次整个延续周期的定期风险评价报告即可。 某II类产品2,其首个注册期是2014年6月至2019年6月。注册人应提交2019年的年度报告,数据汇总期是2018年6月至2019年6月,即上图中红色时间段。 某III类产品3,其首个注册期是2015年6月至2020年6月。该产品需要提交2期定期风险评价报告。分别是上图中绿色时间段和黄色时间段。
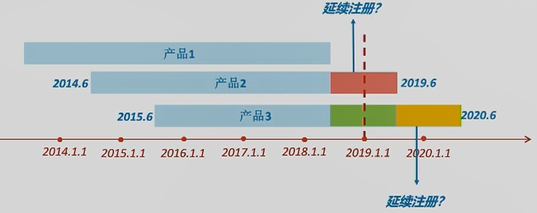
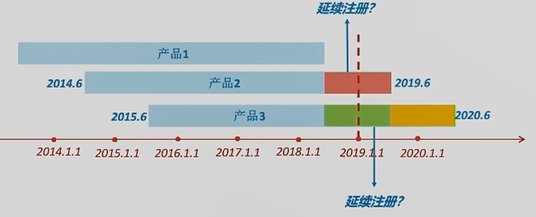
问:若产品2在蓝色箭头时间点进行了延续注册,定期风险评价报告要如何提交呢? 答:这是一种特殊情况。在1号令生效时,该产品已经处于延续注册状态。所以对于这类情况,2019年的年度定期风险评价报告就不用提交了。
问:若产品3在蓝色箭头时间点进行了延续注册,其当期定期风险评价报告要如何提交呢? 答:1号令规定:提前延续注册,仍应按照首个注册期要求,提交当期定期风险评价报告。产品3的当期即为黄色时间段,所以即便其在黄色时间段内延续注册,提交的定期风险评价报告也必须是整个黄色时间段的报告。
问:若产品3在绿色部分就进行了延续注册,其当期定期风险评价报告要如何提交呢? 答:产品3的当期定期风险评价报告的数据汇总期就是整个绿色时间段。黄色时间段的数据,就作为下一次延续注册时,定期风险评价报告中的一部分内容。 06 热点问题 问 产品处于首个注册期内,之前有销售,现已停止生产销售,是否仍需要继续提交定期风险评价报告? 答
是。即便现在停止生产销售,市面上仍会有该产品在流通使用。为了保证该产品的风险可控,还是需要定期按时提交定期风险评价报告。
问 产品取得注册证后从未上市销售是否需要提交定期风险评价报告? 答 是。但在系统内提交定期风险评价报告时,可在报告正文部分做简写和简要说明。或者由公司提交“该产品从未在国内外上市”的说明,来替代报告的正文。
问 应急注册产品是否需要提交定期风险评价报告? 答 尚不需要应急注册产品提交报告,但是应急注册产品转为正式注册以后,需要按照首个注册期的要求,提交定期风险评价报告。同时,第一份评价报告的汇总期应涵盖其应急注册期。 07 总结 《定期风险评价报告》
定位:产品上市后风险管理的一环 特点:精准、全周期、趋势分析 重点:风险(不良事件报告)分析与评价 要点:理解规范要求进行汇总、撰写 |
| 上一条:关于2021年7月~8月医疗器械注册受理前技术问题咨询工作安排的通告(2021年第10号) | 下一条:加强技术审评与注册质量管理体系核查关联 |